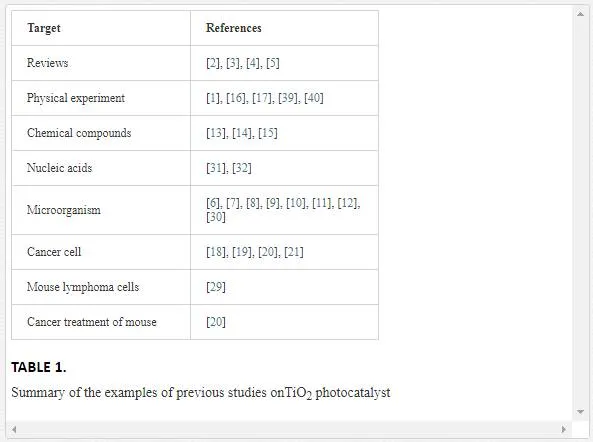
Titanium dioxide (TiO2), a semiconducting material, is a well-known photocatalyst [1-5]. Examples of previous studies about TiO2 photocatalytic reactions are listed in Table 1. A nanoparticle (NP) of TiO2also demonstrates photocatalytic activity. Important applications of TiO2 photocatalysts are bactericidal activity [2-4, 6-12] and degradation of chemical pollutants [2-4, 13]. Related physical and chemical mechanisms have been also investigated [2-5, 14-17]. Photo-irradiated TiO2 NPs induce the formation of various reactive species, leading to the damage of biomacromolecules. These reactive species include hole (h+), either free or trapped hydroxyl radicals (OH⋅), superoxide (O2⋅-), hydrogen peroxide (H2O2), and singlet oxygen (1O2), among others. Hydroxyl radicals, O2⋅-, H2O2, and 1O2 are the typical reactive oxygen species. TiO2 photocatalysts have been found to kill cancer cells [18-21] other than bacteria, viruses, and algae under ultraviolet-A (wavelength: 315–400 nm) illumination [2-4, 6-12]. Therefore, one of the potential applications of the TiO2 NP photocatalyst is photodynamic therapy (PDT), which is a promising treatment for cancer and some nonmalignant conditions [22-25]. In general, the mechanism of cytotoxicity by the photocatalysis of TiO2 is based on cell membrane damage via the generation of the aforementioned reactive oxygen species. Furthermore, DNA damage in human cells [26-28], mouse lymphoma cells [29], and phage [30] by the TiO2 NP photocatalyst has been reported. Direct damage of isolated DNA by TiO2 photocatalyst in vitro has been also studied [31, 32]. However, the DNA-damaging mechanism in vivo is not well-understood, because the incorporation of the TiO2 NPs in the nucleus is difficult [18]. A previous study has shown that H2O2 formation through the photocatalytic reaction of TiO2 may contribute to cellular DNA damage [2, 19].
Hydrogen peroxide, a long-lived reactive oxygen species, can penetrate the nucleus membrane and induce oxidation of the nucleobase and strand breakage through enhancement by metal ions. Iron or copper ions can enhance the activity of H2O2 to produce OH⋅ [33] and copper-peroxide [34-36]. Furthermore, secondary generation of reactive oxygen species may contribute to cytotoxicity of TiO2 NPs photocatalyst [37]. Since the photocatalytic reaction will occur in a complex biological environment, an interaction between TiO2 NPs and biomaterials should participate in the generation of reactive species to induce DNA damage. For example, sugars photocatalyzed by TiO2 NPs may secondarily generate H2O2 through their further oxidation process by molecular oxygen in the presence of a metal ion [37]. In addition, the possibility of 1O2-mediated cytotoxicity by TiO2 NPs has been proposed [38]. Actually, 1O2 generation by photo-irradiated TiO2 NPs was demonstrated by a near-infrared spectroscopy [39, 40]. In this chapter, recent studies about photocatalytic biomacromolecule damage by TiO2 NPs are briefly reviewed.
GENERAL MECHANISM OF PHOTOCATALYSIS OF TIO2 NP
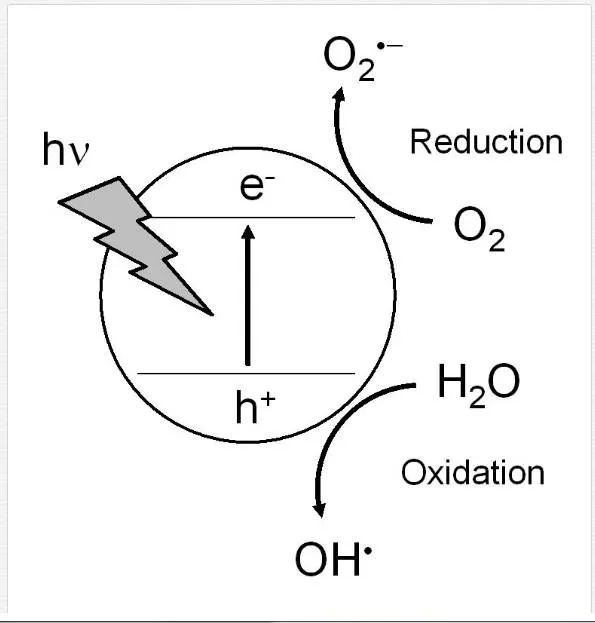
The crystal of TiO2 is a semiconductor, and the two crystalline forms, anatase and rutile, are well-known (Figure 1) [2-5]. The values of the band gap energy of these crystal forms are 3.26 and 3.06 eV for anatase and rutile, respectively. Photo-irradiation to a TiO2 crystal induces the formation of an excited electron (e–) in the conduction band and an h+ in the valence band, leading to the redox reaction of materials adsorbing on the TiO2 surface, including water and/or molecular oxygen. The photocatalytic reactions with its surface water and oxygen cause the formation of various reactive oxygen species such as free or trapped OH⋅, O2⋅-, H2O2, and 1O2 [2-5].

An excited electron in the conductive band reduces the oxygen molecule adsorbed on the surface of TiO2 NPs, leading to the generation of various reactive oxygen species as follows (Figure 2):

The reaction (3) is mediated by ultraviolet radiation (hν, wavelength <355 nm), metal ions (Mn+) such as Fe2+, and O2⋅-, as follows [33]:
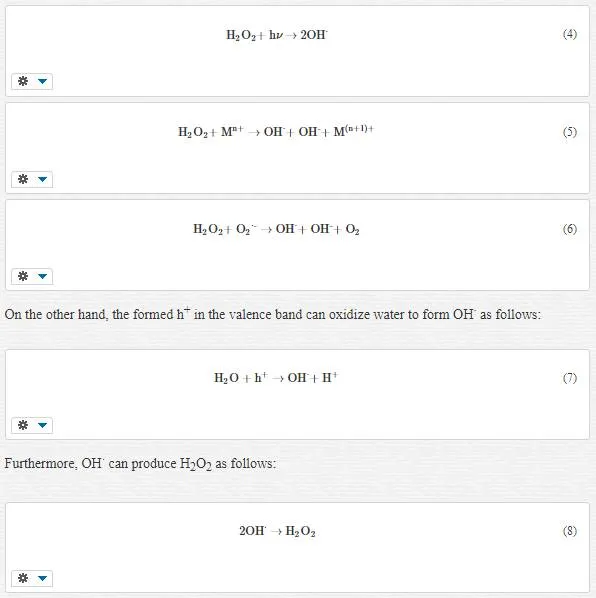

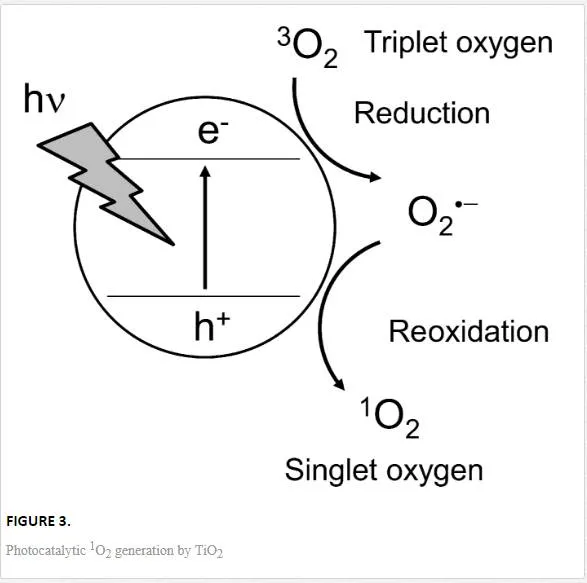
A photo-irradiated TiO2 NP can induce 1O2 formation. The formation of 1O2 is considered to be an important mechanism of PDT. This reaction can be explained by the following process: O2⋅- formed by TiO2 photocatalysis is reoxidized by the h+ of TiO2 on the particle surface to form 1O2 as follows (Figure 3):

These reactive oxygen species should contribute to the mechanism of the phototoxicity induced by TiO2NPs.
STERILIZATION EFFECT BY TIO2
One of the most important medicinal applications of TiO2 NPs is to kill bacteria on its surfaces. TiO2NPs under ultraviolet radiation produce a strong oxidative effect through the formation of above-mentioned reactive oxygen species and can be used as a photocatalytic disinfectant without other chemical reagents. Fujishima and coworkers reported the bactericidal effect of TiO2 photocatalysts against Escherichia coli under ultraviolet-A irradiation using black light [6]. This is the first report of the application of phototoxicity of TiO2 NPs. It was speculated that H2O2 was a reactive species responsible for this phototoxic effect [7]. Relevantly, the photocatalytic effect of TiO2 against methicillin-resistant Staphylococcus aureus (MRSA) and Clostridium difficile in hospitals has been reported [10]. The bactericidal effect of TiO2 NPs could be enhanced by metal doping [9]. Furthermore, visible-light-induced TiO2 photocatalysts were developed and utilized in antibacterial applications. For example, sulfur-doped TiO2 demonstrates the killing effect on Escherichia coli under white-light irradiation commonly used in hospitals [11].
PHOTODYNAMIC THERAPY
Photodynamic therapy, which is a promising and less-invasive treatment for cancer, employs a photosensitizer and visible light to produce oxidative stress in cells and ablate cancerous tumors [22-25]. Photodynamic therapy is also used for treating some nonmalignant conditions that are generally characterized by the overgrowth of unwanted or abnormal cells. In general, porphyrins are used as photosensitizers under visible-light irradiation, since the human tissue has relatively high transparency for visible light, especially red light, and visible light has hardly any side effects. In the case of visible light PDT, 1O2 is considered an important reactive species for PDT because 1O2 can be easily generated by visible light [41-44]. Critical targets of the generated 1O2 include mitochondria and enzyme proteins. Moreover, DNA is also an important target biomolecule of photosensitized reactions [45-49]. Relevantly, photocatalytic 1O2 generation by TiO2 has been reported [38-40].
TiO2, a nontoxic material, is chemically stable, and demonstrates a phototoxic effect. Therefore, an application of TiO2 for PDT has been investigated [2]. The cytotoxicity of an illuminated TiO2 film electrode for HeLa cells [18,19] and T-24 human bladder cancer cells [21] has been reported. Animal experiments also demonstrated the antitumor effect of TiO2 NPs [20]. This report showed an antineoplastic effect on skin cancer in mouse models.
Photo catalytic DNA damage by TiO2 NPs
Cellular DNA damage photocatalyzed by TiO2 NPs was demonstrated by the experiment using cancer cells [18,19,21]. TiO2 NPs can be taken into the cancer cell [27]; however, incorporation into the cell nucleus is difficult [18]. Therefore, it is speculated that the indirect mechanism contributes to DNA damage induced by photo-irradiated TiO2 NPs. Hence, model experiments using isolated DNA were performed [31, 32]. In this section, an example of photocatalytic DNA damage by TiO2 NPs was introduced.
ISOLATED DNA DAMAGE PHOTOCATALYZED BY TIO2 NPS AND ITS SEQUENCE SPECIFICITY
Photo-irradiated TiO2 NPs catalyze DNA damage in the presence of copper(II) ion [31]. Relevantly, copper-aided photosterilization of microbial cells on TiO2 was reported [8]. DNA damage by anatase NPs is more severe than that by rutile NPs. The DNA damage is enhanced by piperidine treatment, because photo-irradiated TiO2 NPs cause not only DNA strand breakage but also base oxidation. In general, hot piperidine cleaves DNA strand at modified base. Photo-irradiated TiO2 NPs induce the formation of piperidine-labile products at the bolded site of 5’-TG, 5’-TG, and 5’-TC (Figure 4). Furthermore, TiO2 NPs photocatalyze DNA strand cleavage at the bolded guanines of 5’-TG and 5’-TCin a DNA fragment treated with E coli formamidopyrimidine-DNA glycosylase (Fpg protein), which can catalyze the excision of piperidine-resistant 8-oxo-7,8-dihydro-2’deoxyguanine (8-oxo-G) [50,51]. The formation of 8-oxo-G was confirmed by an analysis with a high-performance liquid chromatography (Figure 5). In addition, Fpg protein can cleave the oxidized cytosine, such as 5-hydroxy cytosine [52]. These results suggest that photo-irradiated TiO2 NPs induce 8-oxo-G formation adjacent to piperidine-labile thymine lesions. Such double-base lesions should be generated from one radical hit that leads through a secondary reaction to a tandem base damage at pyrimidine and adjacent residues [53-56]. Actually, it has been reported that H2O2 induces tandem mutations in human cells via vicinal or cross-linked base modification in the presence of copper(II) ion [57]. Since repairing of cluster DNA damage in living cells is difficult [58], such clustered base damage, including double-base lesions, appears to play an important role in the phototoxicity of TiO2 NPs.


MECHANISM OF DNA DAMAGE PHOTOCATALYZED BY TIO2 NPS
Catalase, a well-known scavenger of H2O2, and bathocuproines, a copper(I) ion chelator, inhibit DNA damage photocatalyzed by TiO2 NPs, whereas, typical OH⋅ scavenger cannot inhibit the DNA damage. These results suggest that H2O2 and copper(I) ion participate in DNA damage by photo-irradiated TiO2NPs. It has been reported that OH⋅ is not the main reactive species involved in DNA damage by H2O2and copper(I) ions [34-36, 59]. DNA-associated copper(I) ions may generate other oxidants, including a copper–peroxo intermediate, such as Cu(I)-OOH, which is generated from the reaction of H2O2 and copper(I) ions [34-36, 59]. Indeed, methional, which can scavenge Cu(I)-OOH [36, 59], shows inhibitory effect on DNA damage photocatalyzed by TiO2 NPs. The generation of these reactive species may be responsible for the formation of piperidine-labile products and 8-oxo-G.
On the other hand, a high concentration of anatase NPs can catalyze DNA photodamage without copper(II) ions. Typical OH⋅ scavengers, ethanol and sugars, effectively inhibit the DNA photodamage by a high concentration of anatase NPs. The DNA damage induced by photo-irradiated anatase NPs without copper(II) ions is observed at every nucleobases without site specificity. Such DNA damage without sequence-specificity is the typical pattern of OH⋅-mediated DNA damage [34]. A proposed mechanism of DNA damage photocatalyzed by TiO2 NPs is shown in Figure 6. The crystalline forms of TiO2, anatase and rutile, are semiconductors with band gap energies of 3.26 and 3.06 eV, which correspond to the following wavelengths of light: 385 and 400 nm, respectively. When a TiO2 semiconductor NPs absorbs photon with energy greater than their band gap, electrons in the valence band are excited to the conduction band, creating electron-h+ pairs and causing various chemical reactions [2-5]. The electron acts as a reductant, whereas the h+ is a powerful oxidant. In aqueous environments, oxygen molecule can be reduced by the electron into O2⋅-, and water molecule can be oxidized by the h+ into OH⋅. In general, formed O2⋅- can be dismutated into H2O2 by proton. The oxygen reduction may precede the reduction of copper(II) ions under aerobic condition, since the concentration of dissolved oxygen is higher (~250 μM) than that of the copper(II) ion used in this study (20 μM).
The copper(II) reduction may be mediated by O2⋅-. Hydrogen peroxide reacts with copper(I) ions to generate other oxidants, including a copper–peroxo intermediate, resulting in the oxidation of DNA bases. Copper ions, which are essential components of chromatin [60,61], are found to bind DNA with high affinity [62,63]. Therefore, copper ions may play an important role in reactive oxygen generation in vivo, although mammals have evolved means of minimizing the levels of free copper ions and most copper ions bind to protein caries and transporters [64]. Hydroxyl radicals formed by the reaction of water with an h+ in the valence band of TiO2 NPs also slightly participate in DNA damage photocatalyzed by anatase NPs. Because OH⋅ is a strong oxidative agent, OH⋅ can damage every nucleobase [34]. The present results suggest that H2O2 mainly participate in the phototoxicity of TiO2NPs and the contribution of OH⋅ is relatively small. Fujishima et al. also reported the involvement of H2O2 generated from O2⋅- in the cytotoxicity of illuminated TiO2 NPs [2-4, 8-13].

TiO2 NPs might be a potential agent for PDT [22-25]. TiO2 NPs can be incorporated into cancer cells and demonstrate cytotoxicity under photo-irradiation [2-4, 26-28]. Photocatalytic reaction by TiO2 NPs induces a number of functional changes in cell including altered permeability of cellular membranes to potassium and calcium ions, release of RNA and proteins and cytotoxicity [2,18-21]. It has been reported that DNA can be a target biomolecule of the photocatalytic reaction of TiO2 NPs [26-30]. Although incorporation of TiO2 NPs into cell nucleus is difficult [18], the generated H2O2 by a photocatalytic reaction of TiO2 NPs can be easily diffused and incorporated in a cell nucleus, leading to DNA photodamage with metal ions. Relevantly, several studies demonstrated that DNA is a potential target of PDT [47,65,66]. Therefore, the metal-mediated DNA damage through the photocatalysis of TiO2 NPs may participate in cytotoxicity by photo-irradiated TiO2 NPs.
Secondary production of reactive oxygen species from photocatalyzed materials by TiO2 NPs
As mentioned above, DNA damage in human cells by TiO2 NPs has also been reported [26-28]. The direct DNA damage by TiO2 NPs photocatalyst in vitro has been also studied [31, 32]. However, the DNA-damaging mechanism in vivo is not well-understood because the incorporation of the TiO2 NPs in the cell nucleus is difficult [18]. Since the TiO2 photocatalytic reaction occurs in a complex biological environment, an interaction between TiO2 NPs and biomaterials may participate in the generation of reactive species to induce DNA damage. Hence, the effect of sugars, which are ubiquitous biomaterials, on DNA damage photocatalyzed by TiO2 NPs was examined [37].
In the case of anatase, a high concentration of TiO2 NPs can damage DNA at every nucleobase by OH⋅generation in the absence of copper(II) ions. Typical free OH⋅ scavengers inhibited this copper(II)-independent DNA damage. These results indicate that free OH⋅ partly contributes to DNA damage photocatalyzed by TiO2. On the other hand, scavengers of OH⋅, such as a sugar (mannitol), ethanol, and formate, enhanced the copper(II)-dependent DNA damage [31]. These scavengers themselves did not induce DNA damage.
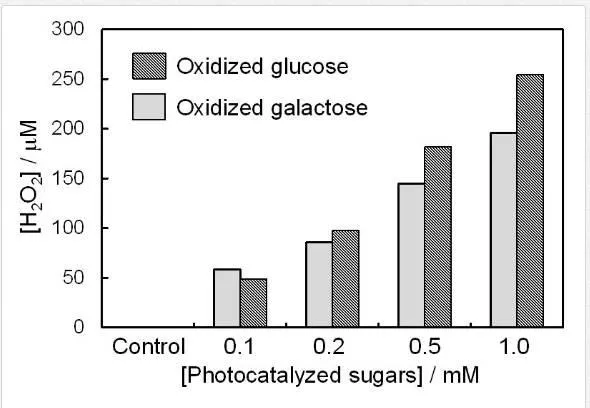
Since OH⋅ can oxidize most biomaterials, the oxidized products of biomaterials by the TiO2 photocatalyst may damage DNA via the generation of secondary reactive oxygen species. The addition of sugars, glucose and galactose, which are ubiquitous biomolecules, enhanced the DNA damage photocatalyzed by TiO2 NPs. Enhancement of DNA damage by sugars has seldom been reported, and these sugars themselves could not induce DNA damage. Therefore, the products of the photocatalytic reaction of these sugars by TiO2 NPs is responsible for the copper(II)-dependent damage to DNA. Indeed, the glucose and galactose oxidized by the TiO2 photocatalytic reaction caused DNA damage in the presence of copper(II) ion [37]. The inhibitory effect of various scavengers for DNA damage by the photo-oxidized products of sugars by TiO2 was examined. Catalase inhibited DNA damage by the photocatalyzed glucose, indicating the involvement of H2O2. Bathocuproine, which is a chelator of copper(I) ion, also inhibited DNA damage by the photocatalyzed glucose, suggesting the involvement of copper(I) ion. The free OH⋅ scavengers had no or little inhibitory effect on DNA damage. The inhibitory effect of superoxide dismutase (SOD) was weak, suggesting that O2⋅- itself is not the main reactive species for DNA damage. Similar results were observed in the case of galactose. Fluorometry using folic acid [67] demonstrated the formation of H2O2 from the photocatalyzed sugars (Figure 7). The amount of H2O2 generation was comparable with that of other H2O2-mediated DNA-damaging drugs [68]. H2O2 generation was not observed in the absence of copper(II) ions. These results showed that the oxidized products of sugars generate H2O2 during the reaction with copper(II) ions, resulting in secondary DNA damage.
These sugars act as an electron donor for the photocatalytic reaction [15,37]. Partially oxidized sugars, such as aldehyde compounds, are possibly produced through this photocatalytic oxidation. The mechanism of DNA damage by the photocatalyzed product of sugars is proposed in Figure 8. Aldehydes can generate H2O2 via its further oxidation [69], though these sugars themselves are stable compounds. Many studies have reported DNA damage by H2O2 and copper(II) ions [34-36, 70]. Various chemical compounds, including aldehydes, easily produce O2⋅- through their autoxidation process. The autoxidation is markedly enhanced by copper(II) ion, which is an essential component of chromatin [60, 61]. The formed O2⋅- is rapidly dismutated into H2O2. Although the generated H2O2itself cannot damage DNA, H2O2 reduces copper(II) into copper(I), leading to the activation of H2O2through the formation of reactive species, such as Cu(I)-OOH [34-36, 59]. Indeed, methional, a scavenger of Cu(I)-OOH, inhibited the DNA damage. This reactive species cannot be scavenged by the free OH⋅ scavengers; however, it can effectively oxidize the nucleobases [34-36, 59].
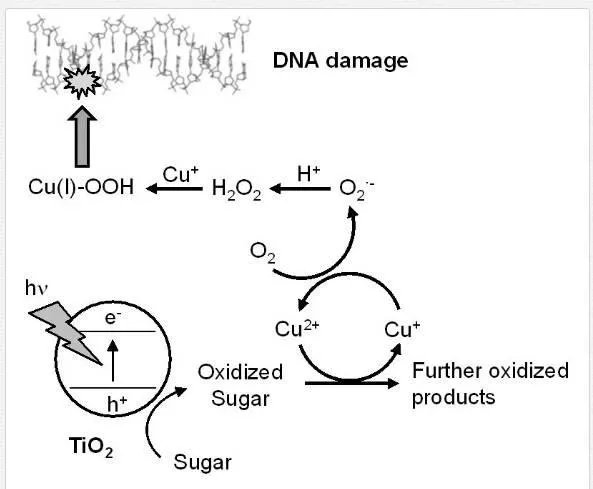
Although TiO2 is not likely to be incorporated in a cell nucleus [18], H2O2 generated via a photocatalytic reaction can be easily diffused and incorporated in a cell nucleus. This DNA-damaging mechanism via H2O2 generation may participate in the phototoxicity of TiO2. In vivo, the cell membrane is an important reaction field for the TiO2 photocatalyst because TiO2 NPs show affinity with a cell membrane [18]. Further, a part of the TiO2 NPs can become incorporated into the cell. Sugars on the cell membrane and cytoplasm may be oxidized by the TiO2 photocatalytic reaction. The generated h+ and OH⋅ can oxidize these sugars, leading to the formation of secondary H2O2 from their photo-oxidized products.
In summary, sugars enhance the DNA damage photocatalyzed by TiO2 NPs. This enhancement of DNA damage is due to the secondary generation of a reactive oxygen species, H2O2, which can diffuse in the cell and damage cellular DNA. These findings suggest that the secondary H2O2 generation contributes to the phototoxicity of TiO2 more than the direct formation of reactive oxygen species does.
Singlet oxygen formation through photocatalytic reaction of TiO2 NPs
A contribution of 1O2 in the TiO2 photocatalytic reaction was reported [38]. Singlet oxygen generation by TiO2 photocatalysis has been demonstrated by the emission measurement of 1O2, which is assigned to the transition from 1O2(1Δg) to 3O2(3∑g) [39, 40]. Because 1O2 is considered to be an important reactive species in PDT process [22-25], the clarification of the contribution of 1O2 generated by TiO2photocatalysis is closely related to a design of photocatalyst for medicinal application. Thus, 1O2generation in the TiO2 photocatalysis and its importance on biomolecular damage was examined [40]. The typical emission of 1O2 at around 1270 nm was observed during irradiation of TiO2 NPs. Relatively strong emission of 1O2 was observed in nonpolar organic solvents such as dichloromethane. The quantum yield (ΦΔ) of 1O2 generation by TiO2 photocatalysis in ethanol was estimated from the comparison of 1O2 emission intensities by TiO2 NPs and methylene blue (ΦΔ= 0.52) [71] and the absorbance of the TiO2 NP dispersions. Because the scattering by suspended TiO2 NPs makes the calculation of absorbed light intensity complex, the precise estimation of the ΦΔ is difficult. Thus, the ΦΔ was estimated using the apparent absorbance of TiO2 NPs. The calculated value indicates the lowest limit of the ΦΔ by TiO2 photocatalysis in ethanol. The reported lifetime of 1O2 generated via TiO2photocatalytic reaction is 5 μs [39]. This value is shorter than that by the photosensitized reaction of methylene blue (12 μs) [72]. Since the emission intensity of 1O2 is proportional to its lifetime, the ΦΔwas corrected by the lifetime of 1O2. The estimated value of ΦΔ by both types of TiO2, anatase and rutile, was about 0.02 in ethanol. This value of ΦΔ is enough large to induce oxidative damage to biomolecules. The 1O2 emission in D2O was completely quenched by the addition of SOD, which is the enzyme to dismutate O2⋅- into H2O2. These results can be explained by the fact that 1O2 is formed by the reoxidation of O2⋅-, generated from the photoreduction of oxygen molecules by TiO2 NPs (Figure 3). The intensity of 1O2 emission observed in the case of rutile was significantly larger than that by anatase in D2O. The difference of the 1O2 generation by these two types of TiO2 crystalline forms can be reasonably explained by that in aqueous solution. H2O2 generation proceeds in the photocatalysis of anatase rather than O2⋅- generation, whereas O2⋅- is the main product from oxygen photoreduction mediated by rutile [17]. These results support the mechanism of 1O2 generation via O2⋅- by TiO2photocatalysis.
The emission spectrum of 1O2 by TiO2 (in both, anatase and rutile type cases) slightly blue-shifted (~4 nm) compared with that by methylene blue. These results suggest that the surroundings of the 1O2generated on the TiO2 surface are different from that by methylene blue in solution. In the case of the photosensitization of methylene blue, the generated 1O2 deactivates in the homogeneous media of solvents. A possible explanation of the blue-shift is that most of the 1O2 generated by TiO2 NPs deactivates on the TiO2 surface. The intensity of 1O2 emission by TiO2 photocatalysis in liposome was significantly larger than that in an aqueous solution in both, anatase and rutile type cases. The enhancement of the 1O2 emission can be explained by the elongation of the lifetime of 1O2 or the acceleration of the photocatalytic reaction. This result shows that phospholipids membrane is an important environment of the phototoxic reaction mediated by 1O2 in the photocatalytic reactions of TiO2 NPs. Indeed, high affinity of TiO2 NPs with a cell membrane was reported [18]. Consequently, an environmental effect of a cell membrane is important for the photocatalytic reaction of TiO2 NPs. Since amino acid residues in proteins can be oxidized by 1O2 [42], a membrane protein should be the target biomolecule in cell membrane. Indeed, 1O2 emission was quenched by the addition of bovine serum albumin, a typical water soluble protein, suggesting scavenging of the 1O2 generated by TiO2 photocatalysis through oxidation of protein.
In vivo, nicotinamide adenine dinucleotide (NADH) is one of the most important target biomolecule oxidized by 1O2 [73, 74]. NADH demonstrates the typical absorption peak at around 340 nm in an ultraviolet absorption spectral measurement, and this absorption band is diminished by the oxidation. It has been reported that TiO2 NPs hardly induce the oxidation of NADH in aqueous solution during ultraviolet irradiation. Since NADH hardly adsorbed on a surface of TiO2 NPs, the 1O2 could not effectively oxidize NADH in solution. As mentioned above, it has been reported that photo-irradiated TiO2 NPs can induce DNA damage mainly through H2O2 and OH⋅, and the 1O2-mediated DNA damage could not be observed [31]. These reports concluded that the photocatalytic 1O2 generation on the surface of TiO2 NPs is not important in the damaging mechanism of the biomolecules such as DNA and NADH, of which the affinity with TiO2 surface is small.
In conclusion, photo-irradiated TiO2 NPs can produce 1O2 through reoxidation of O2⋅-, which is formed by photocatalytic reduction of oxygen molecule on the surface of TiO2 NPs. Since most of the 1O2deactivated on TiO2 surface, the 1O2 on TiO2 surface cannot induce the oxidation of DNA and NADH. However, the 1O2 generation by TiO2 photocatalysis could be enhanced in the microenvironment of phospholipids membrane. These findings suggest that 1O2 may contribute to phototoxicity of TiO2 NPs through oxidation of membrane protein.
Conclusions
TiO2 NPs photocatalyze DNA oxidation. A relatively small concentration of TiO2 NPs frequently induces tandem base oxidation at guanine and thymine residues through H2O2 generation in the presence of a copper(II) ion. A copper–peroxo complex is considered to be an important reactive species responsible for this DNA damage. In addition, cytosine residues are also photooxidized by TiO2NPs. In the case of a high concentration of TiO2 NPs, OH⋅ contributes to DNA damage without sequence specificity. In the presence of sugars, TiO2 NPs indirectly induce DNA damage by the secondary H2O2, which is produced through an autoxidation process of the photo-oxidized products of sugars by TiO2 NPs. Furthermore, 1O2 is also produced by photo-irradiated TiO2 NPs. The 1O2generation is explained by the reoxidation of O2⋅-, which is produced by photocatalytic reduction of the oxygen molecule adsorbed on the surface of TiO2 NPs. The photocatalyzed formation of 1O2 might contribute to the oxidation of the membrane protein. These mechanisms of photocatalytic reactive oxygen formation should be involved in the photocytotoxicity of TiO2 NPs. Because TiO2 is a chemically stable and nontoxic material, the bactericidal activity and cytotoxicity against cancer cells will play more important roles in the field of medical applications of nanomaterials.